山东能源研究院/青岛能源所提出微生物组功能的校正算法Meta-Apo
基于元基因组测序的微生物组功能分析和比较在疾病诊断、生态监控、生物安全等领域具有广阔应用价值,但是高昂的成本限制了其更广泛应用。日前,青岛能源所单细胞中心开发了微生物组功能校正算法Meta-Apo(Metagenomic Apochromat),为大规模的菌群功能比较提供了一种保证分析精度的同时可大幅降低实验和计算成本的解决方案。
目前,微生物组功能的分析和比较,主要基于基因组序列进行代谢重建。路线之一是鸟枪法元基因组测序(Whole Metagenome Sequencing;WMS),然而其高昂的测序成本(~1000~2000元/样本)和冗杂的分析过程阻碍了该方法的大规模应用。另一方面,通过扩增子测序(~200元/样本),利用16S rRNA等进化标记基因和其参照基因组之间的关联,亦能推断微生物组的功能。该方法虽然大幅降低了成本,但16S rRNA基因片段经常带有扩增偏好性,而且与全基因组序列的关联并非完全可靠,因此,该方法的功能重建结果经常会出现较大的偏差。
为了解决以上问题,单细胞中心生物信息研究组提出了Meta-Apo算法,通过挖掘WMS数据和16S rRNA扩增子数据之间的同构关系,利用少量WMS和16S rRNA数据对(即同一个菌群样本分别进行WMS测序和16S rRNA扩增子测序)进行训练,来实现对大规模16S rRNA扩增子测序样本的菌群功能校正(图1)。研究结果显示,Meta-Apo利用仅仅15例样本的数据对进行训练,进而基于16S rRNA扩增子对5,000例人体肠道菌群样本进行功能预测和校正,其结果与同批样本基于WMS推断的菌群功能基本一致,而总测序成本仅为后者的20%左右。因此,针对大规模菌群样本的功能重建这一目的,将全部样本用16S rRNA扩增子策略,同时用WMS策略测定其中的一小部分样本,将能在保证分析精度的前提下、大幅降低实验和计算成本。因此,Meta-Apo算法和相应的测序策略为大规模菌群功能分析项目的设计提供了一个重要的工具。
该项工作由单细胞中心与青岛大学计算机科学技术学院合作完成,该论文第一作者是生物信息研究组荆功超助理研究员,通讯作者是苏晓泉教授,并获得了国家自然科学基金、山东省自然科学基金、中国博士后科学基金的支持。(文/图 荆功超)
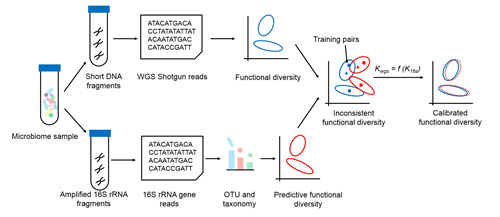
图1 Meta-Apo算法能校正基于16S基因扩增子推断的菌群功能
原文链接:
Gongchao Jing, Yufeng Zhang, Wenzhi Cui, Lu Liu, Jian Xu, Xiaoquan Su*. Meta-Apo improves accuracy of 16S-amplicon-based prediction of microbiome function. BMC Genomics 22, 9 (2021). https://doi.org/10.1186/s12864-020-07307-1
附件下载: